 https://www.genetica.cz
+420 272 701 055
Služeb 4
Praha
https://www.genetica.cz
+420 272 701 055
Služeb 4
Praha





4.png)


2.png)
Customer siccess stories::
Rapid teljes genom szekvenálás (rWGS)
Projekt leírás
A koncepció bizonyítása nyomán végzett komplex rWGS szekvenálási projektet Prof. Milan Macek, a prágai Károly Egyetem 2. Orvostudományi Kara és a Motol Egyetemi Kórház Biológiai és Orvosi Genetikai Tanszékének vezetőjének csapatával együttműködve végeztük. A mintákat – 3 fős család, szülők és Mayer-Rokitansky-Küster-Hauser (MRKH) szindrómával diagnosztizált proband - Prof. Macek csapata választotta ki, annak érdekében, hogy demonstrálják a teljes genom szekvenálásban (WGS) rejlő lehetőséget és az alkalmazás hatékonyságát a különböző panel és/vagy WES elemzéssel szemben.
Emellett a teljes folyamatot élőben közvetítették a március 20-21-én Lipno-ban megrendezett ’DNA diagnostika 2023’ konferencián.
Bevezetés
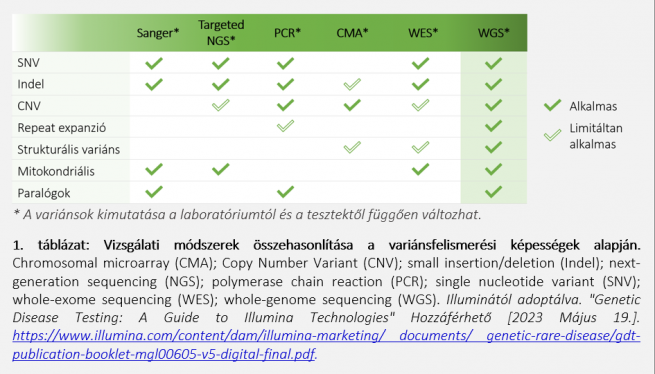
A rapid teljes genom szekvenálás (rWGS) áttörést jelentő megközelítés, különösen azokban az esetekben, amikor a pontosság mellett a sebesség is kulcsfontosságú tényező a komplex genomelemzésben. Kiváló szemléltető példa erre a gyermekkori/újszülöttkori vizsgálat, mivel az újszülöttkori halálozások 40%-a ritka, rendkívül gyorsan lezajló genetikai betegségekkel kapcsolatosak.1-4 A klinikai gyakorlatban a rWGS segíthet a kezelés hatékonyságának javításában vagy a betegség lefolyásának prognózisának meghatározásában azáltal, hogy más diagnosztikai megközelítésekkel összehasonlítva (1. táblázat) gyorsan azonosítja a betegségben szerepet játszó genetikai variánsokat.5
Ezen túlmenően, a rWGS a gyors személyre szabott diagnosztika kivételesen magas értékű megoldásává és úgynevezett arany standardjává válik - előnyeihez viszonyítva kifejezetten alacsony költséggel jár és az ok-okozati variánsok korai és pontos azonosításával többek között az egészségügyi ellátás összköltségének csökkentését eredményezi.6
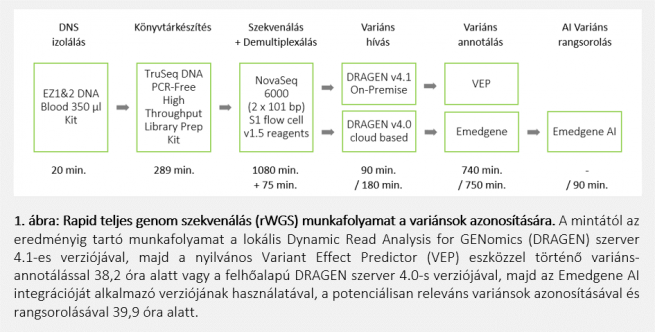
Az Institute of Applied Biotechnologies a.s. (IAB) intézettel együttműködve bemutatunk egy magas minőségű kimenettel rendelkező gyors WGS protokollt, amely a QIAGEN és az Illumina NGS technológiáira és eszközeire épül. Az optimalizált NGS protokoll a legfejlettebb termékeket és megközelítéseket ötvözi a DNS-izolálás, a könyvtárkészítés, a szekvenálás és az adatelemzés, majd az annotált variánsok értelmezése során. Az rWGS protokollnak és Prof. Maceknek köszönhetően a vizsgált trió (a proband és szülei) eredményei fenomenális 39,9 óra alatt kerültek feldolgozásra és kiértékelésre (1. ábra).
Módszerek
DNS tisztítás: A feldolgozási idő drámai csökkenése már a teljes munkafolyamat kezdeti lépése során is biztosított az EZ2 Connect (QIAGEN) automatizált mágneses gyöngy alapú tisztítás alkalmazásával. A trió 3 kis térfogatú vérmintájából mindössze 20 percen belül kiváló minőségű genomi DNS-t (gDNS) izoláltunk az EZ1&2 DNS Blood 350 µl-es kit (QIAGEN) segítségével.
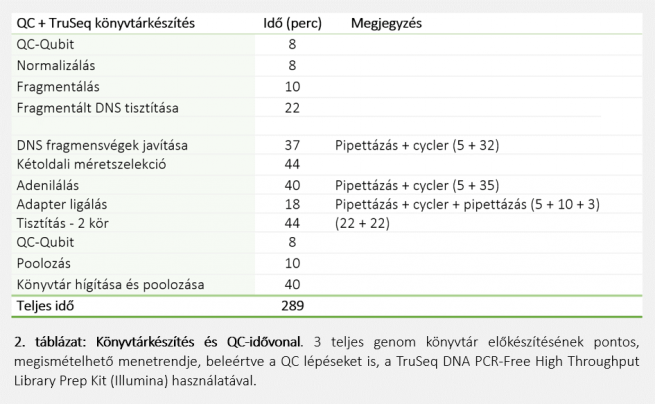
Könyvtárkészítés: A családi egyes tagjainak teljes genom könyvtárait 1050 ng koncentrációjú gDNS-ből kiindulva 289 perc alatt készítettük el, hígítottuk és denaturáltuk, beleértve az összes minőségellenőrzésre irányuló lépést is (2. táblázat), a TruSeq DNA PCR-Free High Throughput Library Prep Kit (Illumina) segítségével. Az alkalmazott, PCR lépéseket nem tartalmazó protokoll kiváló megoldást nyújt a nehezen szekvenálható DNS-régiók, például GC-gazdag régiók, promóterek vagy ismétlődő szakaszok számára. Emellett a mechanikus fragmentálás alkalmazása csökkenti a nem kívánt artefaktumok és a könyvtárakban keletkező hézagok kockázatát.
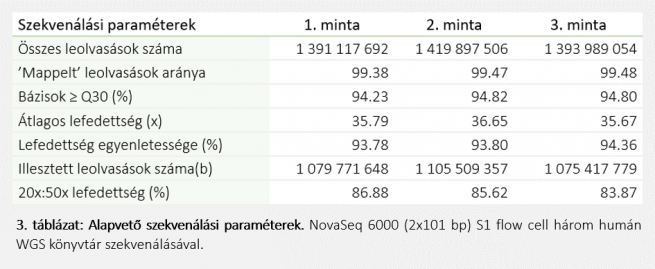
Szekvenálás: A trió rWGS szekvenálását NovaSeq 6000 rendszerrel végeztük S1 flow cell (2x101 bp) (Illumina) alkalmazásával. A szekvenálási kísérletet 18 óra alatt sikeresen elvégeztük, 464,92 Gb adatkimenettel, 92,57%-ban 35x-nél nagyobb lefedettséggel és 94,66%-ban 30-nál nagyobb Phred minőségi pontszámmal (Q30). Mindhárom teljes genom könyvtár hozta az előírt 800 millió párosított végű olvasás határértéket (3. táblázat).
Adatelemzés: A szekvenálási adatok demultiplexelése, a CBCL FASTQ-vá történő konvertálásával a BaseSpace Sequence Hub felhőalapú szerverén (Illumina) 75 percen belül történt. A másodlagos elemzés downstream lépéseihez két különböző megközelítést alkalmaztunk.
Az első esetben a lokális Dynamic Read Analysis for GENomics (DRAGEN) On-Premise Server v4.1 (Illumina) segítségével hg38-as referencia genomra való illesztést és Variáns hívást, majd a nyílt forráskódú Variant Effect Predictor (VEP) annotációs eszköz (Ensembl) segítségével ~14 óra alatt létrehozta az annotált variánsok listáját. Ez az internetes feltöltési sebességkorlátozások hiányának és a feldolgozási prioritások teljes ellenőrzésének köszönhető. A felhőalapú DRAGEN v4.0 és Emedgene (Illumina) rendszereken futó másik rendelkezésre álló opció ~15,5 óra alatt végezte el a referencia genomra való illesztéstől a variáns annotálásig tartó folyamatot.
Ez a kisebb időbeli késedelem azonban nagyon hamar megtérül, amikor az elemzőknek szelektálniuk kell az Emedgene mesterséges intelligenciája (AI) által előzetesen kiválasztott néhány klinikailag releváns variánst a több száz rendezetlen annotált variáns helyett.
Ezesetben, az Emedgene AI 7 legvalószínűbb variánst és 63 további lehetséges variánst választott ki, amelyeket a Károly Egyetem 2. Orvostudományi Karának és a Motol Egyetemi Kórház laboratóriumi diagnosztikusaihoz küldtek el elemzésre. Az esetek mélyebb feltárásához az Emedgene rendelkezik egy gyors szűrési funkcióval, amely lehetővé teszi a klinikusok számára, hogy a jelölt gének exon- és intronváltozatait megtekintsék, vagy az Emedgene-fiókjukon belül a várható szegregációval rendelkező változatokra összpontosítsanak.
Összefoglalás
Ez a rövid ismertető egy úttörő rWGS protokollról számol be, amely különböző digitális genomikai módszereket, köztük mesterséges intelligenciát integrál egy átfogó személyre szabott diagnosztikai megközelítésbe, hogy a mintagyűjtéstől számított kevesebb mint 40 órán belül rangsorolt, annotált variánsokat szolgáltasson. A QIAGEN és az Illumina csúcstechnológiáin alapuló, a pontosságot, a sebességet és a kivételes analitikai komplexitást ötvöző innovatív protokollunk teljesen új szintre emeli a személyre szabott diagnosztikát, és megnyitja a humángenetika klinikai gyakorlatban történő felhasználásának lehetőségeit. A bemutatott eredmények azt mutatják, hogy az rWGS kétségtelenül az egyik legfejlettebb precíziós diagnosztikai megoldás, amely fantasztikus eredményeket mutat a genetikai rendellenességek okozati variánsainak gyors és pontos kimutatásában, ahol más diagnosztikai módszerek kudarcot vallanak.
Hatalmas gratuláció és köszönet illeti elsősorban prof. Milan Macek-et és csapatát, az IAB-t, illetve a GeneTiCA csapatából mindazokat, akik közreműködtek az egész projekt megvalósításában!
_______________________________________________________________________________________
1. Weiner J, Sharma J, Lantos J, Kilbride H. How infants die in the neonatal intensive care unit: trends from 1999 through 2008. Arch Pediatr Adolesc Med. 2011;165(7):630-634. doi:10.1001/archpediatrics.2011.102
2. Gunne E, McGarvey C, Hamilton K, Treacy E, Lambert DM, Lynch SA. A retrospective review of the contribution of rare diseases to paediatric mortality in Ireland. Orphanet J Rare Dis. 2020;15(1):311. doi:10.1186/s13023-020-01574-7Weiner
3. Berry MA, Shah PS, Brouillette RT, Hellmann J. Predictors of mortality and length of stay for neonates admitted to children's hospital neonatal intensive care units. J Perinatol. 2008;28(4):297-302.
4. Arth AC, Tinker SC, Simeone RM, Ailes EC, Cragan JD, Grosse SD. Inpatient Hospitalization Costs Associated with Birth Defects Among Persons of All Ages - United States, 2013. MMWR Morb Mortal Wkly Rep. 2017;66(2):41-46.
5. Petrikin JE, Cakici JA, Clark MM, et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom Med. 2018;3:6. doi:10.1038/s41525-018-0045-8
6. Farnaes L, Hildreth A, Sweeney NM, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med. 2018;3:10. doi:10.1038/s41525-018-0049-4